
HOME
>
試薬
>
細胞情報伝達
>
エピジェネティクス
>
ヒストン脱修飾酵素(Eraser)関連化合物
HOME
>
試薬
>
創薬研究
>
探索研究
>
スクリーニング/探索用商品
>
ヒストン脱修飾酵素(Eraser)関連化合物
ヒストン修飾の研究に有用なツール ヒストン脱修飾酵素(Eraser)関連化合物
掲載日情報:2025/10/07 現在Webページ番号:65523

ヒストン修飾は、クロマチン構造の変化をさせることでエピジェネティックな遺伝子発現制御に関与しています。ヒストン修飾を付加する“Writer”、除去する“Eraser”、修飾されたヒストンを認識するタンパク質“Reader”が関与し、細胞の分化・発達を調節しています。本ページでは、各種ヒストン修飾酵素(Eraser)を阻害する化合物などをご紹介いたします。
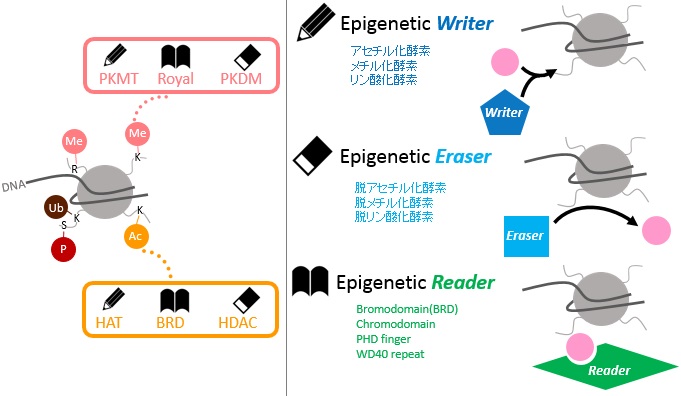
追加しました。
ヒストンの各種修飾による機能の解析法
化学プローブによる解析
化学プローブは、明確な作用機序をもち、細胞内で標的タンパク質の活性を強力かつ高選択的に調節する、十分に特性評価された低分子化合物(阻害物質・活性化物質・分解誘導化合物など)を指します。理想的な化学プローブの特徴として、力価はIC50またはKd値が100 nM未満を目標とし、同ファミリー内で30倍以上の選択性を確保しつつ、オフターゲット低減を確認できるものとされています。
PROTACによる解析
PROTACは、2つの化学構造が連結したヘテロバイファンクショナル分子です。1つは目的の標的タンパク質と結合し、もう1つはE3ユビキチンリガーゼと結合し、ユビキチンタグの付加とそれに続くプロテアソーム分解が起こります。
追加しました。
ヒストンのリジンメチル化
ヒストンのリジンメチル化は、メチル化部位に応じて転写活性に関与する場合と転写抑制に関与する場合があります。最もよく研究されているヒストンのリジンメチル化部位は、ヒストンH3のK4(H3K4)、K9(H3K9)、K27(H3K27)、K36(H3K36)、K79(H3K79)およびヒストンH4のK20(H4K20)です。
ヒストンリジン脱メチル化酵素(KDM)
ヒストンリジン脱メチル化酵素は、メチル化されたリジンからメチル基を除去する反応を触媒します。KDM1ファミリーは、リジンからモノメチル化およびジメチルされたアミノ酸残基を脱メチル化するのに対し、KDM2~8のファミリーはJumonji C(JmjC)ドメインを含み、モノメチル化、ジメチル化、さらにトリメチル化されたアミノ酸残基を脱メチル化します。特定KDMの過剰発現は、さまざまながんにおいて観察されています。
KDM1阻害物質
KDM1A(LSD1)およびKDM1B(LSD2)は、H3K4me1/me2の脱メチル化を触媒します。またKDM1Aは、H3K9me1/me2の脱メチル化も触媒します。
商品コードをクリックすると価格表をご覧いただけます。
| 品名 | 商品コード |
|---|---|
| GSK2879552 | 19403 |
| GSK-LSD1(hydrochloride) | 16439 |
| ORY-1001 | 19136 |
| SP-2509 | 15487 |
KDM4(JMJD2)ファミリー阻害物質
KDM4(JMJD2)ファミリーは、H3K9me2/me3またはH3K36me2/me3の脱メチル化を触媒します。
商品コードをクリックすると価格表をご覧いただけます。
| 品名 | ターゲット | 商品コード |
|---|---|---|
| KDM4D-IN-1 | JMJD2D | 37431 |
| ML-324 | JMJD2E | 17472 |
| NCGC00244536 | JMJD2B | 18478 |
| N-Oxalylglycine | JMJD2A、JMJD2C、JMJD2E | 13944 |
| Prohexadione | JMJD2A | 33922 |
KDM5(JARID1)ファミリー阻害物質
KDM5(JARID1)ファミリーは、H3K4me2/me3の脱メチル化を触媒します。
商品コードをクリックすると価格表をご覧いただけます。
| 品名 | ターゲット | 商品コード |
|---|---|---|
| CPI-455 | JARID1A-D | 22127 |
| Esculetin | JARID1B | 19286 |
| JQKD82(hydrochloride) | JARID1A | 41542 |
| PBIT | JARID1A~D | 16272 |
KDM6ファミリー阻害物質
KDM6ファミリーは、H3K27me2/me3の脱メチル化を触媒します。
商品コードをクリックすると価格表をご覧いただけます。
| 品名 | ターゲット | 商品コード |
|---|---|---|
| GSK-J1(sodium salt) | JMJD3、UTX | 12054 |
| GSK-J4(hydrochloride) | JMJD3、UTX | 12073 |
Multi/Pan Jumonji阻害物質
商品コードをクリックすると価格表をご覧いただけます。
| 品名 | ターゲット | 商品コード |
|---|---|---|
| Caffeic Acid | JMJD2C、UTX | 70602 |
| Daminozide | KDM2A、KDM7A、PHF8 | 12033 |
| IOX1 | 2-oxoglutarate oxygenases、including JMJD2A、JMJD2C、JMJD2E、KDM3A、JARID1C、JMJD3 | 11572 |
| JIB-04 | Pan-Jumonji(JARID1A、JMJD2A、JMJD2B、JMJD2C、JMJD2E、JMJD3) | 15338 |
| Methylstat(hydrate) | JMJD2A、JMJD2C、JMJD2E、JMJD3、PHF8 | 11091 |
| PFI-90 | JKDM3B、JMJD2B、JARID1A、JMJD3 | 40120 |
| TC-E 5002 | KDM2A、KDM7A、PHF8 | 17717 |
追加しました。
ヒストンのアセチル化
ヒストンのアセチル化は、リジン残基の正電荷を中性化し、クロマチン構造の弛緩と転写活性化およびDNA損傷応答に関与しています。
ヒストン脱アセチル化酵素(HDAC)
ヒストン脱アセチル化酵素は、ヒストン上のアセチル化リジン残基から負に帯電したアセチル基を除去する酵素です。その結果、クロマチン構造が凝縮し、ヒストンとDNAの結合が強固になり、転写抑制されます。ClassⅠ、Ⅱ、ⅣはZn2+依存性HDAC(HDAC1~11)で構成されるのに対し、ClassⅢはNAD+依存性Sirtuin(SIRT1~7)で構成されます。HDAC阻害物質はT細胞リンパ腫、多発性骨髄腫、デュシェンヌ型筋ジストロフィー(DMD)の治療薬としてFDA承認されています。
| ClassⅠ | HDAC1 | HDAC2 | HDAC3 | HDAC8 | |||
|---|---|---|---|---|---|---|---|
| ClassⅡa | HDAC4 | HDAC5 | HDAC7 | HDAC9 | |||
| ClassⅡb | HDAC6 | HDAC10 | |||||
| ClassⅢ | SIRT1 | SIRT2 | SIRT3 | SIRT4 | SIRT5 | SIRT6 | SIRT7 |
| ClassⅣ | HDAC11 |
ClassⅠ阻害物質
商品コードをクリックすると価格表をご覧いただけます。
| 品名 | ターゲット | 商品コード |
|---|---|---|
| MS-275 | HDAC1、HDAC3 | 13284 |
| PCI 34051 | HDAC8 | 10444 |
| RGFP966 | HDAC3 | 16917 |
| Romidepsin | ClassⅠHDAC | 17130 |
| Valproic Acid(sodium salt) | ClassⅠHDAC | 13033 |
ClassⅡ阻害物質
商品コードをクリックすると価格表をご覧いただけます。
| 品名 | ターゲット | 商品コード |
|---|---|---|
| ACY-241 | HDAC6 | 26173 |
| ACY-1215 | HDAC6 | 21531 |
| LMK 235 | HDAC4、HDAC5 | 14969 |
| MC 1568 | ClassⅡa HDAC | 16265 |
| Nexturastat A | HDAC6 | 16874 |
| TMP195 | ClassⅡa HDAC | 23242 |
| TMP269 | ClassⅡa HDAC | 17738 |
| Tubastatin A | HDAC6 | 15785 |
ClassⅢ活性化物質/阻害物質
商品コードをクリックすると価格表をご覧いただけます。
■ SIRT活性化物質
| 品名 | ターゲット | 商品コード |
|---|---|---|
| BML-278 | SIRT1、SIRT2、SIRT3 | 20209 |
| MDL 800 | SIRT6 | 35528 |
| SRT 1720(hydrochloride) | SIRT1 | 10011020 |
| SRT 2104 | SIRT1 | 28380 |
| trans-Resveratrol | SIRT1 | 70675 |
■ SIRT阻害物質
商品コードをクリックすると価格表をご覧いただけます。
| 品名 | ターゲット | 商品コード |
|---|---|---|
| 3-TYP | SIRT3 | 29660 |
| AGK2 | SIRT2 | 13145 |
| (±)-EX-527 | SIRT1 | 10009798 |
| Nicotinamide | SIRT1 | 11127 |
| OSS-128167 | SIRT6 | 29052 |
| SirReal2 | SIRT2 | 18116 |
| Sirtinol | SIRT1、SIRT2 | 10523 |
| Tenovin-1 | SIRT1、SIRT2 | 13085 |
Multi/Pan HDAC阻害物質
商品コードをクリックすると価格表をご覧いただけます。
| 品名 | 商品コード |
|---|---|
| Belinostat | 34084 |
| ITF 2357 | 11045 |
| Mocetinostat | 18287 |
| Panobinostat | 13280 |
| SAHA | 10009929 |
| Sodium Butyrate | 13121 |
| Sulforaphane | 10496 |
| Trichostatin A | 89730 |
HDAC/SIRT活性物質/阻害物質スクリーニングアッセイキット
品名をクリックすると各製品の詳細、商品コードをクリックすると価格表をご覧いただけます。
■ HDACアッセイキット
| 品名 | 商品コード |
|---|---|
| HDAC Cell-Based Activity Assay Kit | 600150 |
| HDAC Fluorometric Activity Assay Kit | 10011563 |
| HDAC1 Inhibitor Screening Assay Kit | 10011564 |
| HDAC8 Inhibitor Screening Assay Kit | 700230 |
■ SIRTアッセイキット
追加しました。
関連製品
品名をクリックすると各製品の詳細、商品コードをクリックすると価格表をご覧いただけます。
| 分類 | 品名 | 商品コード |
|---|---|---|
| アッセイキット | LSD1 Inhibitor Screening Assay Kit | 700120 |
| 化合物ライブラリー | Epigenetics Screening Library | 11076 |
| 化学プローブセット | SGC Epigenetic Probe Set | 17748 |
追加しました。
製品情報は掲載時点のものですが、価格表内の価格については随時最新のものに更新されます。お問い合わせいただくタイミングにより製品情報・価格などは変更されている場合があります。
表示価格に、消費税等は含まれていません。一部価格が予告なく変更される場合がありますので、あらかじめご了承下さい。