
HOME>
試薬>
細胞情報伝達>
エピジェネティクス>
Cayman Chemical社 エピジェネティクス関連低分子化合物
ヒストン脱修飾酵素(Eraser)関連化合物
HOME>
試薬>
創薬研究>
探索研究>
スクリーニング/探索用製品>
Cayman Chemical社 エピジェネティクス関連低分子化合物
ヒストン脱修飾酵素(Eraser)関連化合物
Eraser関連低分子化合物
Cayman Chemical社 エピジェネティクス関連低分子化合物
ヒストン脱修飾酵素(Eraser)関連化合物
掲載日情報:2017/04/03 現在Webページ番号:65523

Check it out!
Epigenetics Screening Library
エピジェネティクスにおいて、”writer”、”eraser”、”reader”として知られる約140種の低分子化合物のライブラリー「Epigenetics Screening Library」の詳細については、こちらをご覧下さい。

Epigenetics Screening Library (#11076)
追加しました。
低分子化合物製品
項目をクリックすると一覧表がご覧いただけます。
| Eraser | Reader | Writer |
|---|
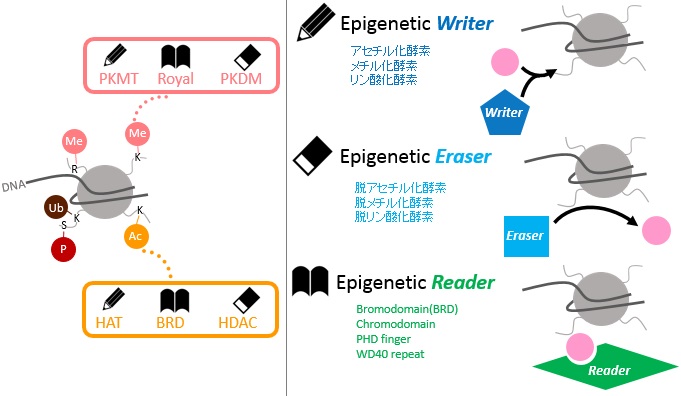
細胞ごとにクロマチンの状態は、さまざまな修飾によって制御されています。修飾を担う酵素“Writer”、修飾されたヒストンを認識するタンパク質“Reader”、脱修飾する酵素“Eraser”が関与し、細胞の分化・発達を調節しています。
参考文献
William P. Janzen, et al., Drug Discovery Today: Technologies, 7(1), (2010).
Alexander Tarakhovsky, Nature Immunology, 11, 565~568 (2010).
追加しました。
Eraser関連化合物
商品コードをクリックすると価格表がご覧いただけます。
| 商品コード | 品名 | 製品詳細 |
|---|---|---|
| 15205 | 2-Hexyl-4-Pentynoic Acid | Inhibits HDAC activity much more potently (IC50=13μM) than valproic acid (IC50=398μM); induces histone hyperacetylation in cerebellar granule cells significantly at 5μM; induces the expression of Hsp70-1a and Hsp70-1b and protects cerebellar granule cells from glutamate-induced excitotoxicity |
| 10495 | 4-iodo-SAHA | A hydrophobic derivative of the class I and class II HDAC inhibitor SAHA that demonstrates >60% inhibition of HDAC1 and HDAC6 activity in a deacetylase activity assay; inhibits proliferation of SK-BR-3 breast-derived, HT29 colon-derived, and U937 leukemia cell lines with EC50 values of 1.1, 0.95, and 0.12μM, respectively |
| 13856 | 5-Nitroso-8-quinolinol | An HDAC inhibitor, antitumor agent that at 10μM can induce oxidative stress contributing to apoptosis and differentiation in MCF-7 breast cancer cells; inhibits T. gondii tachyzoite propagation in human fibroblasts (EC50=80 nM) and inhibits P. falciparum growth in human red blood cells (EC50=1.3μM) |
| 10575 | Apicidin | A fungal toxin that demonstrates selective inhibition of HDAC3/NcoR over HDAC6 (IC50=15.8 and 665.1 nM, respectively); has broad spectrum activity against Apicomplexan parasites and exhibits antiproliferative activity against various cancer cell lines (IC50=0.13-2.36μM) |
| 19834 | BRD4884 | An HDAC inhibitor (IC50=29 nM, 62 nM, and 1.09μM for HDAC1, 2, and 3, respectively); possesses preferential binding kinetics with seven-fold longer half-life on HDAC2 compared to HDAC1 (143 min vs. 20 min) |
| 19836 | BRD6688 | An HDAC inhibitor (IC50=21 nM, 100 nM, and 11.48μM for HDAC1, 2, and 3, respectively); possesses six6-fold extended half-life for binding on HDAC2 compared to HDAC1 (381 min vs. 65 min); rescues the memory defects associated with p25 induced neurodegeneration in contextual fear conditioning in a CK-p25 mouse model of neurodegeneration |
| 89740 | CAY10398 | An inhibitor of HDAC (IC50=10μM) |
| 10005019 | CAY10433 | An HDAC inhibitor with an IC50 value of 30μM when tested in HeLa cell nuclear extracts using 200μM acetylated fluorometric substrate |
| 13146 | CAY10603 | A potent and selective inhibitor of HDAC6 (IC50=0.002 nM, as compared with 271, 252, 0.42, 6851, and 90.7 nM for HDAC1, 2, 3, 8, and 10, respectively); prevents the growth of several pancreatic cancer cell lines (IC50=0.1-1μM) |
| 15403 | CAY10683 | A potent HDAC inhibitor that inhibits HDAC2 and HDAC6 with IC50 values of 0.119 and 434 nM; ineffective against HDAC4 (IC50=>1,000 nM); inhibits the growth of HCT116 cells and HuT-78 cells (GI50=29.4 and 1.4μM, respectively) more effectively than human dermal fibroblasts (GI50=>100μM) |
| 13172 | CBHA | HDAC1 and HDAC3 inhibitor (ID50=0.01 and 0.07μM, respectively, in vitro); induces apoptosis in nine different neuroblastoma cell lines in culture (0.5-4.0μM) and completely suppresses neuroblastoma tumor growth in SCID mice at 200 mg/kg |
| 13686 | Chidamide | An HDAC inhibitor that increases histone H3 acetylation levels in LoVo and HT-29 colon cancer cells at concentrations as low as 4μM; dose-dependently decreases the activation of several oncogenic signaling kinases and induces cell cycle arrest in colon cancer cells |
| 12084 | CI-994 | An inhibitor of class I HDACs with IC50 values of 0.9, 0.9, 1.2, and >20μM for recombinant human HDAC 1, 2, 3, and 8, respectively; displays a wide spectrum of antitumor activity, particularly in tumors normally refractory to conventional anticancer agents |
| 10671 | Coumarin-SAHA | A fluorescent probe that competitively binds HDAC; demonstrates fluorescence excitation and emission maxima of 325 and 400 nm, respectively, which is quenched by 50% when bound to HDAC |
| 16426 | CUDC-101 | A multi-target inhibitor that potently blocks EGFR and HER2 (IC50=2.4 and 16.4 nM, respectively) and inhibits the activity of class I and class II HDACs at nanomolar concentrations (e.g., IC50=4.5, 12.6, 13.2, and 11.4 nM for HDAC1, 2, 4, and 5, respectively) |
| 10576 | HC Toxin | A cell-permeable, reversible inhibitor of HDACs (IC50=30 nM) |
| 15200 | HDAC6 Inhibitor | A potent and selective inhibitor of HDAC6 (IC50=36 nM) that poorly blocks other HDAC enzymes; cell permeable, inhibiting the acetylation of tubulin in cells with an IC50 value of 210 nM |
| 13295 | HNHA | A cell-permeable inhibitor of HDAC activity (IC50=100 nM) |
| 15066 | HPOB | A potent, selective inhibitor of HDAC6 (IC50=56 nM); induces acetylation of α-tubulin but not histones; enhances the cytotoxicity of the broad spectrum HDAC inhibitor SAHA against cancer cells in nude mice carrying an androgen-dependent CWR22 human prostate cancer xenograft |
| 11045 | ITF 2357 | Inhibits maize HDACs: HD2, HD-1B, and HD-1A (IC50 =7.5-16 nM) and reduces TNFα, IL-1α, and IL-1β production (IC50=10-22 nM); inhibits activity of cells expressing JAK2V617F (IC50=1-10 nM), which are implicated in the pathophysiology of many myeloproliferative diseases |
| 18681 | ITSA1 | A cell-permeable suppressor of TSA action, reducing TSA-induced acetylation of histone and tubulin in A549 cells when given at 50μM; prevents TSA-mediated cell cycle arrest and apoptosis; blocks the action of the HDAC inhibitor SAHA, preventing SAHA-induced hyperacetylation of tubulin in A549 cells |
| 14088 | JNJ-26481585 (hydrochloride) | A hydroxamate-based HDAC inhibitor that shows activity toward all HDAC enzymes with highest potency in vitro toward HDAC1 (IC50=0.11 nM) |
| 16427 | LAQ824 | A hydroxamate-based inhibitor of HDACs with an IC50 value of 30 nM; inhibits the growth of colon, breast, prostate, and non-small cell lung cancer cell lines at concentrations of less than 1μM; augments the actions of fludarabine and imatinib mesylate in human leukemia cells |
| 14969 | LMK 235 | An HDAC inhibitor that selectively targets HDACs 4 and 5 (IC50=12 and 4 nM, respectively) over other HDACs (IC50=56, 320, 850, 880, and 1,280 for HDACs 6, 1, 11, 2, and 8, respectively); displays enhanced cytotoxic effects against human cancer cell lines, compared to SAHA or TSA |
| 13174 | M 344 | An inhibitor of HDACs, inhibiting maize HDAC (IC50=100 nM) as well as human HDAC1 (IC50=46 nM); shows a 3-fold selectivity for HDAC6 over HDAC1 |
| 18288 | MI-192 | An HDAC inhibitor that preferentially inhibits HDAC2 (IC50=30 nM) and HDAC3 (IC50=16 nM) over HDAC1, 4, 6, 7, and 8 (IC50=4.8, 5, >10, 4.1, and >10μM, respectively).{29271} At 0.1-0.4μM, it induces differentiation and promotes apoptosis of acute myeloid leukemic cell lines U937, HL60, and Kasumi-1 |
| 18287 | Mocetinostat | An orally available HDAC inhibitor that selectively targets HDAC1 and 2 (IC50=0.15 and 0.29μM, respectively), less potently inhibits HDAC3 and 11 (IC50=1.66 and 0.59μM, respectively); induces hyperacetylation of histones, causes cell cycle blockade, and induces apoptosis in cancer cells in vitro |
| 13284 | MS-275 | An inhibitor of HDACs that preferentially inhibits HDAC1 (IC50=300 nM) over HDAC3 (IC50=8μM); does not inhibit HDAC8; induces p21/CIP1/WAF1, slowing cell growth, differentiation, and tumor development in vivo |
| 13176 | Oxamflatin | A potent inhibitor of HDACs (IC50=15.7 nM); has been shown to alter the expression of several genes whose products are involved in cell morphology, motility, apoptosis, and cell cycle control, reducing the proliferation of cancer cells |
| 13280 | Panobinostat | A potent inhibitor of all HDACs, with Ki values ranging from 0.6 to 31 nM for HDAC1-11; leads to acetylation of a range of cellular proteins, resulting in cell cycle arrest and apoptosis in cancer cells; has potential applications in spinal muscular atrophy and HIV therapy |
| 20059 | PCI 24781 | |
| 10444 | PCI 34051 | A potent HDAC8 inhibitor (IC50=0.01μM) with >200-fold selectivity over HDAC isoforms 1, 2, 3, 6, and 10 (IC50=4, >50, >50, 2.9, and 13μM, respectively); induces caspase-dependent apoptosis in cell lines derived from T-cell lymphomas or leukemias (GI50s=2.4 - 4μM) |
| 13212 | Pimelic Diphenylamide 106 | A slow, tight-binding inhibitor of class I HDACs, progressively binding HDACs and remaining bound after wash-out; inhibits class I HDACs (IC50=150, 760, 370, and 5,000 nM for HDAC1, 2, 3, and 8, respectively) but not class II HDACs (IC50 > 180μM for HDAC4, 5, and 7) |
| 13870 | Pyroxamide | An inhibitor of HDAC, including HDAC1 (IC50=0.1-0.2μM); induces growth suppression and cell death of certain types of cancer cells in culture |
| 17553 | Resminostat (hydrochloride) | An orally bioavailable inhibitor of HDAC1, HDAC3, and HDAC6 (IC50=43-72 nM), resulting in hyperacetylation of histone H4 in multiple myeloma cells; abrogates cell growth and strongly induces apoptosis in multiple myeloma cells (IC50=2.5-3μM); dose-dependently inhibits HDAC activity in vivo |
| 17130 | Romidepsin | A natural bicyclic depsipeptide that, following reduction, selectively inhibits class I HDACs (IC50=53, 39, 53, and 26 nM, for HDACs 1, 2, 3, and 8, respectively); has anti-cancer activities, particularly against certain T cell lymphomas |
| 10009929 | SAHA | An HDAC inhibitor of class I and class II HDACs at around 50 nM; arrests cell growth in a wide variety of transformed cells in culture at 2.5-5.0μM |
| 10675 | SAHA-BPyne | A SAHA derivative with a benzophenone crosslinker and an alkyne tag to be used for profiling HDAC activities in proteomes and live cells; labels HDAC complex proteins both in proteomes at 100 nM and in live cells at 500 nM; IC50=~3μM for inhibition of HDAC activity in HeLa cell nuclear lysates in an HDAC activity assay |
| 10443 | SB939 | A pan-HDAC inhibitor (IC50=77 nM in an in vitro HDAC1 activity assay) that prevents proliferation of ovarian (A2780), colon (COLO 205 and HCT116), and prostate cancer (PC-3) cell lines at IC50 values of 0.48, 0.56, 0.48, and 0.34μM, respectively; binds all HDAC isozymes with similar affinity (Kis=16-28 nM) with the exception of HDAC6 and 7 (Kis=247 and 104 nM, respectively) |
| 10572 | Scriptaid | An HDAC inhibitor that has an optimal effective concentration of 6-8μM in a cell-based assay, is less toxic than trichostatin A, and works in a wide variety of biological systems; induces cell cycle arrest in cancer cells in vitro and in vivo; facilitates the cloning of inbred mouse strains produced by somatic cell nuclear transfer |
| 11323 | Sodium 4-Phenylbutyrate | A chemical chaperone that rescues the trafficking of misfolded proteins and weakly blocks HDAC activity (IC50=0.4 mM), leading to cell cycle arrest, differentiation, and/or apoptosis of various tumors |
| 13121 | Sodium Butyrate | A short chain fatty acid that inhibits HDACs, induces growth arrest, differentiation and apoptosis in cancer cells, and suppresses inflammation by reducing the expression of pro-inflammatory cytokines |
| 13168 | Splitomicin | A small molecule inhibitor of Sir2p HDAC activity, displaying higher activity in vivo (minimal inhibitory concentration=0.49μM) than in vitro (IC50=60μM); has diverse effects on mammalian cells |
| 10574 | Suberohydroxamic Acid | A competitive HDAC inhibitor that inhibits HDAC1 (IC50=0.25μM) and HDAC3 (IC50=0.30μM); causes cell differentiation, cell cycle arrest, or apoptosis |
| 10496 | Sulforaphane | An isothiocyanate that potently induces chemopreventative enzymes via Keap1-Nrf2 signaling and ARE-driven gene expression; at 15μM, inhibits class I and II HDAC activity and suppresses tumor growth selectively in cancerous prostate epithelial cells without affecting normal cells |
| 17692 | Tasquinimod | An orally-active quinoline-3-carboxamide that inhibits tumor angiogenesis and supplements radiation or chemotherapy in animal models of prostate cancer |
| 17738 | TMP269 | A cell-permeable inhibitor of class IIa HDACs (IC50=126, 80, 36, and 19 nM for HDAC4, 5, 7, and 9, respectively) |
| 89730 | Trichostatin A | A potent, reversible inhibitor of HDAC (IC50=70 mM) |
| 20220 | Trichostatin C | A glycosylated derivative of trichostatin A that has been shown to induce the differentiation of a mouse erythroleukemia cell line and to increase histone H4 acetylation in B cells, though at higher concentrations than trichostatin A |
| 13691 | Tubacin | A selective HDAC6 inhibitor (IC50=4 nM) used as a biochemical tool to control microtubule-dependent intracellular trafficking, to manipulate the aggresome formation of misfolded proteins in certain diseases, and to study the dynamics of cellular adhesion |
| 15785 | Tubastatin A | Potent HDAC6 inhibitor (IC50=15 nM) with 1,000-fold selectivity against all other HDAC isoforms (IC50 > 16μM), excluding HDAC8 (IC50=0.9μM); displays dose-dependent neuronal protection of primary cortical neuron cultures at 5-10μM |
| 10559 | Tubastatin A (trifluoroacetate salt) | Potent HDAC6 inhibitor (IC50=15 nM) with 1,000-fold selectivity against all other HDAC isoforms (IC50 >16μM), excluding HDAC8 (IC50=0.9μM); induces α-tubulin hyperacetylation at 2.5μM in primary cortical neuron cultures; displays dose-dependent neuronal protection of primary cortical neuron cultures at 5-10μM |
| 13033 | Valproic Acid (sodium salt) | An analog of valeric acid, long used as an anticonvulsant; inhibits Class I HDACs with an IC50 of ~2 mM; also inhibits GSK3 and depletes cellular IP3 |
| 14314 | Plumbagin | A natural 1,4-naphthoquinone that has diverse effects in cells and animals; causes the generation of ROS and induces apoptosis in cancer cells; activates signaling through Nrf2 and ARE, inducing the expression of Nrf2 target genes, including NQO1 and HO-1 in cultured neuronal cells |
| 11138 | 2,4-Pyridinedicarboxylic Acid | Structurally mimics 2-OG and chelates zinc; blocks the activity of 2-OG oxygenases (certain lysine demethylases and hydroxylases); inhibits several Jumonji domain-containing lysine demethylases at low micromolar concentrations and prolyl hydroxlase 1 (IC50=1.5μM) |
| 12033 | Daminozide | A selective inhibitor of the human 2-oxoglutarate (JmjC) histone demethylases KDM2A, PHF8, and KDM7A (IC50=1.5, 0.55, and 2.1μM, respectively) |
| 12054 | GSK-J1 (sodium salt) | A potent, cell impermeable inhibitor of the H3K27 histone demethylases JMJD3 and UTX (IC50=18 and 56μM, respectively as measured by mass spectrometry; IC50=60 nM in JMJD3 antibody-based assays) |
| 12056 | GSK-J2 (sodium salt) | A pyridine regio-isomer of GSK-J1 which poorly inhibits JMJD3 (IC50 > 100μM), making it an appropriate negative control for in vitro studies involving GSK-J1 |
| 12073 | GSK-J4 (hydrochloride) | An ethyl ester prodrug of the JMJD3 selective histone demethylase inhibitor GSK-J1; reduces LPS-induced proinflammatory cytokine production, including that of TNFα (IC50=9μM) in human primary macrophages |
| 12074 | GSK-J5 (hydrochloride) | A pyridine regio-isomer of the JMJD3 inhibitor GSK-J4; cell-permeable and hydrolyzed to a free base, which is a weak inhibitor of JMJD3 (IC50 > 100μM), making it an ideal negative control molecule |
| 11572 | IOX1 | A broad-spectrum inhibitor of 2OG oxygenases that inhibits JMJD2 demethylase activity (IC50=87μM); inhibits JMJD2A, JMJD2E and the 2OG oxygenases PHF8, PHD2, and FIH (IC50=1.7, 2.4, 13.3, 14.3, and 20.5μM, respectively) |
| 15338 | JIB-04 | A pyridine hydrazone that broadly inhibits Jumonji histone demethylases (IC50 values are 230, 340, 435, 445, 855, and 1100 nM for JARID1A, JMJD2E, JMJD2B, JMJD2A, JMJD3 and JMJD2C, respectively); inhibits Jumonji demethylase activity, alters gene expression, and blocks viability of cancer cells both in vitro and in vivo |
| 17472 | ML-324 | A cell-permeable inhibitor of JMJD2E (KDM4DL; IC50=920 nM); reduces the expression of immediate early genes of HSV (IC50=~10μM) and hCMV, blocks viral infection, and suppresses the level of viral reactivation in a ganglia explant model of latently infected mice |
| 13944 | N-Oxalylglycine | A cell permeable inhibitor of α-ketoglutarate-dependent enzymes, including JMJD2A, JMJD2C, and JMJD2E (IC50=250, 500, and 24μM, respectively); inhibits the prolyl hydroxylase domain-containing proteins PHD1 and PHD2 with IC50 values of 2.1 and 5.6μM, respectively |
| 16272 | PBIT | A reversible, cell-permeable inhibitor of JARID1 family demethylases (IC50=6, 3, 4.9, and 28μM for JARID1A, JARID1B, JARID1C, and JARID1D, respectively); increases trimethylation of H3K4 in HeLa cells and blocks the proliferation of tumor cells expressing high levels of JARID1B |
| 10010494 | 2-PCPA (hydrochloride) | An irreversible, mechanism-based inhibitor of lysine-specific demethylase 1 (LSD1) with an IC50 value of 20.7μM and a Ki value of 242.7μM that effectively inhibits histone demethylation in vivo; irreversibly inhibits monoamine oxidases (MAO) A and MAO B with IC50 values of 2.3 and 0.95μM and Ki values of 101.9 and 16μM, respectively |
| 19403 | GSK2879552 | A selective, orally bioavailable, mechanism-based inactivator of LSD1/CoREST activity; enhances H3K4 methylation, increasing the expression of tumor-suppressor genes and preventing growth of AML cell lines (EC50s=2-240 nM) |
| 16439 | GSK-LSD1 (hydrochloride) | An irreversible, mechanism-based inhibitor of LSD1 (IC50=16 nM); induces gene expression changes in various cancer cell lines, inhibiting their proliferation (EC50s <5 nM) |
| 17471 | OG-L002 | A potent inhibitor of LSD1 (IC50=0.02μM) that less effectively inhibits MAO-A and MAO-B (IC50=1.38 and 0.72μM); blocks the expression of IE genes of HSV and hCMV in mammalian cells; represses HSV primary infection in mice and blocks HSV reactivation from latency in a mouse ganglion explant model |
| 19136 | ORY-1001 | An orally available, selective inhibitor of LSD1 (IC50 < 20 nM); targets AML stem cells |
| 18124 | RN-1 (hydrochloride) | A potent, irreversible inhibitor of LSD1 (IC50=70 nM); penetrates the blood-brain barrier and blocks long-term but not short-term memory in mice |
| 15487 | SP2509 | A reversible inhibitor of LSD1 (IC50=13 nM); allows increased methylation of H3K4, driving increased expression of p21, p27 and CCAAT/enhancer binding protein α in cultured AML cells; improves survival alone and synergizes with panobinostat in improving survival of mice engrafted with AML cells |
| 13145 | AGK2 | A cell-permeable, selective inhibitor of SIRT2 (IC50=3.5μM) that minimally affects either SIRT1 or SIRT3; rescues dopamine neurons from α-synuclein toxicity in both in vitro and in vivo Parkinson’s disease models |
| 14004 | AK-7 | A cell- and brain-permeable inhibitor of SIRT2 (IC50=15.5μM); dimishes neuronal cell death induced by mutant huntingtin fragment in culture; down-regulates cholesterol biosynthetic gene expression and reduces total cholesterol levels in neurons in vivo |
| 10009797 | CAY10591 | An activator of SIRT1 that decreases TNF-α levels from 325 pg/ml (control) to 104 and 53 pg/ml at 20 and 60μM, respectively; exhibits a significant dose-dependent effect on fat mobilization in differentiated adipocytes |
| 10009796 | CAY10602 | Derived from HTS for compounds that increase the SIRT1-mediated deacetylation of a SIRT1-specific substrate; dose-dependently suppresses the NF-κB-dependent induction of TNF-α by LPS in THP-1 cells, with approximately 75% inhibition achieved at 60μM, without cytotoxicity |
| 10009798 | EX-527 | A cell-permeable, selective inhibitor of SIRT1 (IC50=98 nM); inhibits other SIRTs only at much higher concentrations and has no effect on other HDACs |
| 19771 | Inauhzin | A cell-permeable, selective SIRT1 inhibitor (IC50=0.7-2μM) that reactivates p53 by inhibiting SIRT1 deacetylation activity; inhibits cell proliferation by inducing p53-dependent apoptosis in various human cancer cells (IC50=5.4, 51.9, 3.2, 33.9, and 85.4μM for H460, H1299, A549, HT-29, and WI38 cells, respectively), as well as in xenograft tumors |
| 14648 | JFD00244 | An inhibitor of SIRT2 (IC50=56.7μM); induces granulocytic differentiation in the acute promyelocytic leukemia cell line NB4 |
| 10641 | JGB1741 | A SIRT1-specific inhibitor (IC50=15μM); inhibits metastatic breast cancer MDA-MB 231 cell proliferation (IC50=512 nM), dose-dependently increasing p53 acetylation and p53-mediated apoptosis in these cells |
| 13178 | Salermide | An inhibitor of SIRT1 and SIRT2, causing tumor-specific apopoptic cell death; causes 90% apoptosis within 72 hours (IC50 ~ 20μM) by reactivating proapototic genes that are repressed by SIRT1 in MOLT4 leukemia cells |
| 18116 | SirReal2 | An aminothiazole that acts as a SIRT-rearranging ligand to selectively inhibit SIRT2 (IC50=140 nM) without effect on SIRT1 or SIRT3-6; increases α-tubulin acetylation in HeLa cells |
| 14407 | SIRT1/2 Inhibitor IV | A cell-permeable inhibitor of SIRT1 (IC50=56μM) and SIRT2 (IC50=59μM); less effectively inhibits SIRT5 (IC50 >300μM) and has no effect on class I and II HDACs; sensitizes H460 lung cancer cells to etoposide and paclitaxel; blocks a SIRT1-dependent hypoxic response in vivo |
| 10523 | Sirtinol | A cell-permeable inhibitor of sirtuin NAD+-dependent deacetylases, inhibiting the yeast sirtuin Sir2p with an IC50 value of 68μM and the human sirtuins SIRT1 and SIRT2 with IC50 values of 131 and 38μM, respectively; does not alter HDAC1 activity |
| 13085 | Tenovin-1 | A small molecule activator of p53 that decreases the growth of BL2 Burkitt’s lymphoma and ARN8 melanoma cells; inhibits the deacetylase activity of purified human SIRT1 and SIRT2 |
| 13086 | Tenovin-6 | A analog of tenovin-1; elevates p53 activity in MCF-7 cells at 10μM and reduces growth of ARN8 melanoma xenograft tumors in SCID mice at a dose of 50 mg/kg |
| 20534 | (+)-Biotin 4-Amidobenzoic Acid (sodium salt) | A substrate of biotinidase, releasing PABA, which can be quantified by either fluorescent or colorimetric methods |
| 11076 | Epigenetics Screening Library (96-Well) | The Epigenetics Screening Library contains more than 140 small molecules that are known to modulate the activity of a variety of epigenetic 'writers and erasers' and “reader” proteins in a 96-well Matrix tube rack format as 10 mM stocks in DMSO. It may include compounds that modulate the activity of methyltransferases, demethylases, histone acetyltransferases, histone deacetylases, and acetylated histone binding proteins. |
| 11931 | GSK4112 | A synthetic agonist for REV-ERBα (EC50=0.4μM) that mimics the action of heme; at 10μM inhibits the expression of the circadian target gene bmal1 and reduces glucose output by 30% in mouse primary hepatocytes by repressing the expression of several gluconeogenic genes |
| 11573 | IOX2 | Potent, cell permeable inhibitor of PHD2 (IC50=21 nM) with over 100-fold selectivity compared to inhibition of JMJD2A, JMJD2C, JMJD2E, JMJD3, or the 2OG oxygenase FIH (IC50 > 100μM); inhibits HIF-1α hydroxylation in RCC4 cells at 50μM |
| 18181 | IOX4 | A selective, potent inhibitor of PHD2 (IC50=1.6 nM); active in vivo, inhibiting prolyl hydroxylation and increasing HIF1α levels in cells (IC50 values range from 5.6 to 11.7μM) and inducing HIF1α and HIF2α expression in mice; penetrates the blood-brain barrier to induce HIF expression in the brain |
| 13956 | S-(5'-Adenosyl)-L-Methionine Chloride (hydrochloride) | A ubiquitous methyl donor involved in a wide variety of biological reactions, including those mediated by DNA and protein methyltransferases |
| 16288 | Tenovin-3 | |
| 16374 | α-Hydroxyglutaric Acid (sodium salt) | An α-hydroxy acid, overproduced in 2-hydroxyglutaric aciduria; mutations in IDH1 and IDH2 cause these enzymes to convert isocitrate to 2-hydroxyglutarate; competitively inhibits α-ketoglutarate-dependent dioxygenases, including lysine demethylases and DNA hydroxylases |
追加しました。
製品情報は掲載時点のものですが、価格表内の価格については随時最新のものに更新されます。お問い合わせいただくタイミングにより製品情報・価格などは変更されている場合があります。
表示価格に、消費税等は含まれていません。一部価格が予告なく変更される場合がありますので、あらかじめご了承下さい。